Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a progressive and life-threatening neuromuscular disorder affecting motor neurons in both the brain and spinal cord. Depending on the site disease onset, patients will first exhibit either weakness in the limbs (spinal onset) or difficulties in swallowing or speaking (bulbar onset).
Despite a large variability in the disease progression and severity, this neurodegenerative disease will evolve irreversibly within three to five years from diagnosis towards a breathing muscle weakness leading to death.
Although the sporadic form is predominant, familial ALS occurs by 5-10% of cases.
Pathophysiology
Environmental factors and genetic susceptibility appear to be critical in ALS pathogenesis.
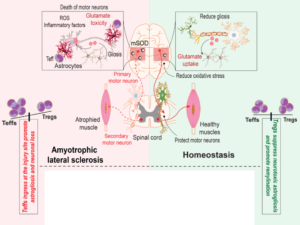
The ALS development is mediated by multiple mechanisms as protein misfolding and aggregation, glutamate toxicity, mitochondrial dysfunction creating a favourable environment for the pro-inflammatory components recruitment, triggering an apoptotic cascade and creating an amplified and self-perpetuating inflammation conducting to neuronal death (Machhi et al. 2020).
Besides, a correlation between Treg and disease severity has been shown. Indeed, Tregs increase during the early slow progressive disease stage whereas a decreased level of Tregs along with a proinflammatory signals increase are reported at the later rapidly accelerating disease stage.
Epidemiology
ALS is the most common adult-onset motor neuron disease and is usually diagnosed between the ages of 40 and 70 with an average age of 55. This disease is approximately 1.5 times more common in men that it is in women.
In the US, approximately 20,000 patients are living with ALS and 5,000 people are newly diagnosed per year.
In Europe, ALS affects about 35,000 patients and causes 10,000 deaths per year. The incidence is ranging from 2.1 to 3.8 per 100 000 person-years.
Disease management
Despite large use of Riluzole (standard of care) and Edaravone (only approved in US and Canada), there is a critical unmet need for effective and safe disease-modifying therapies. Indeed, the former prolongs the survival by 3 months whereas the latter slows the functional decline in patients with ALS with no proven efficacy on survival.
Altogether, the current therapeutic options for treating ALS are not satisfying with 50% of patients dying 2 to 3 years after they have been diagnosed. The need for more efficient drugs with sustained beneficial effect is critical.
What does ILTOO Pharma bring?
ILT-101 is administered as early as possible in the course of the disease. By selectively expanding and activating Treg, ILT-101 is intended to reinstate cerebral homeostasis and inhibit neuroinflammation and self-amplifying loop of neuron cell death. Ultimately, ILT-101 may prolong the survival of ALS patients.